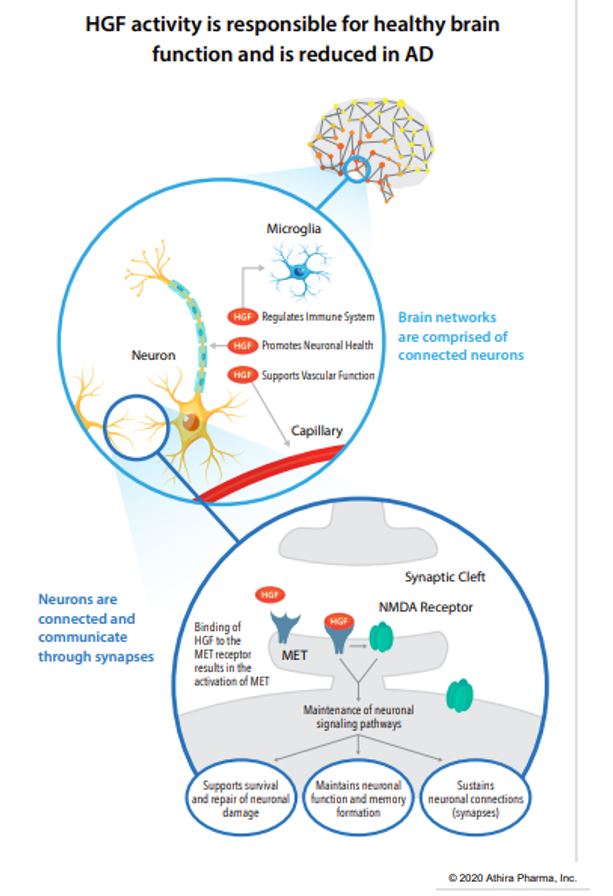
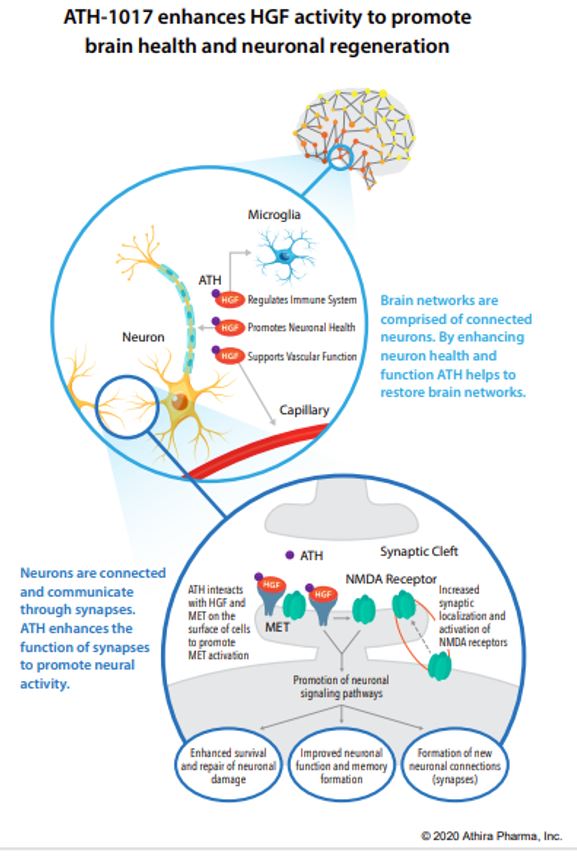
For quite a while, one of the most common methods of isolating disease-risk-associated genes has been a Genome Wide Association Study (GWAS) which involves genotyping a large number of people and associating various genetic loci, or locations, with the phenotypes, or visible traits, that commonly arise from variance at these loci. However, researchers recently developed an even more informative way of performing these genomic analyses, using it to discover 10 new genes that may modify risk of Alzheimer’s disease (AD)! This new method is very similar to GWAS but adds another layer of data regarding protein function, becoming a Proteome-Wide Association Study (PWAS).
PWAS expands upon the information provided by GWAS by analyzing the type of mutation present and quantifying the change in production or functionality of the protein produced by that gene in comparison to controls. Next, protein function scores are correlated to the phenotypes of subjects with those genes to confirm the expected effects. In analysis of a binary phenotype (a trait that is either present or absent with no intermediate presentation) a strong correlation is derived when subjects with a disorder have a significantly different functional effect score than controls, confirming PWAS’s prediction that the protein is less (or more) functional in the mutated form.
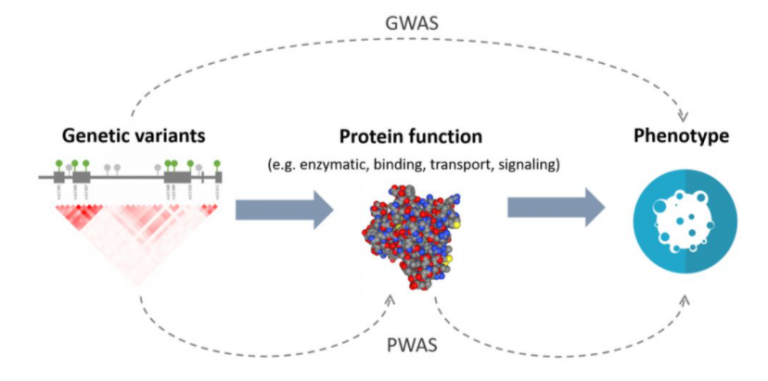
Figure 1. A diagram depicting the difference in analyses between Genome-Wide Association Studies and Proteome-Wide Association Studies.
The additional information provided by PWAS allows detection of associations that are not detectable by GWAS. Researchers at Emory University recently took advantage of this, using PWAS to discover 10 new genes associated with AD risk. To begin, they isolated 1,475 genes whose abundance is genetically controlled and analyzed their AD-risk score in a GWAS dataset (71,880 cases, 383,378 controls). Of the 1,475 genes, only 13 were related to AD risk in the GWAS dataset.
After further analyses with PWAS, including causality tests and adjustments for APOE status, 11 genes remained with evidence for causality of AD. Only 1 of these genes, ACE, had previously been strongly correlated with AD but the other 10 are relatively new discoveries! Those genes include syntaxin 4 and 6, DOC2A, SNX32, ICA1L, cathepsin H, CARHSP1, LACTB, RTFDC1, and PLEKHA1. Although little is known about their relationship to AD pathogenesis, researchers will undoubtedly begin researching how these genes relate to AD risk and development. In time you may even see treatments emerging targeting these genes or proteins.
Even more interestingly, several of these genes impact molecular pathways that are not widely considered part of the disease model for AD, such as LACTB which is a mitochondrial protein (with mitochondrial dysfunction only getting attention as a possible mechanism of AD development within the last few years) and PLEKHA1 which mediates transmembrane signaling more generally. This suggests that there are as-of-yet undiscovered or under-researched factors that relate to AD risk and pathogenesis, opening the door for new treatments and a more complete understanding of the disease itself.