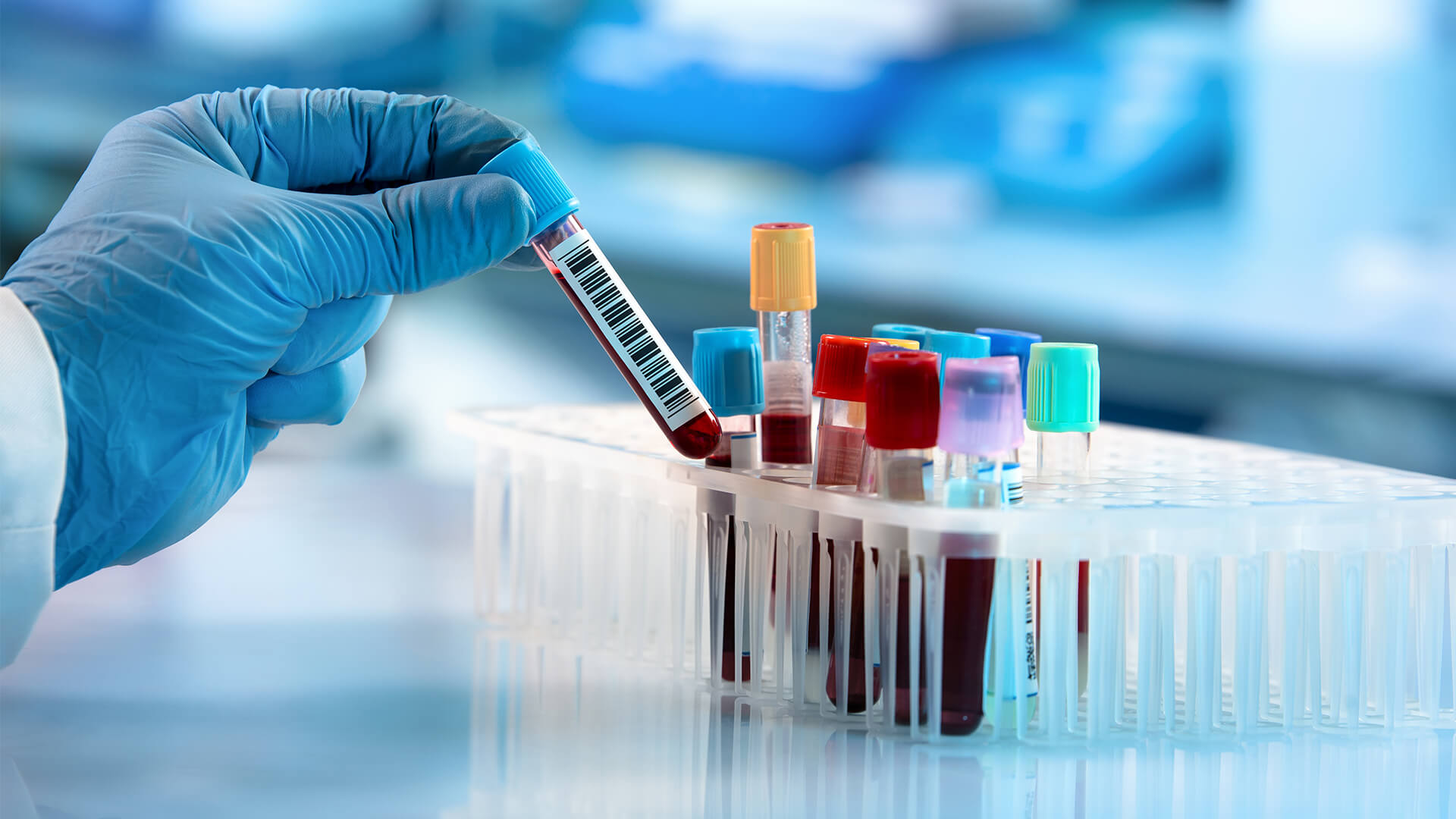
Previously we have discussed
the utilization of blood tests for diagnosing Alzheimer’s Disease (AD). There
are few ways AD can be diagnosed with certainty (i.e., PET scan to assess
amyloid-β, lumbar puncture to assess
cerebrospinal fluid Amyloid and p-tau), these tests can be very expensive or
invasive. Blood tests, on the other hand, are much safer, easier, and of lower
cost. In this blog, we will continue to discuss the promise of blood testing for
AD and its utilization in detecting different stages of the disease through serum
levels of p-tau181.
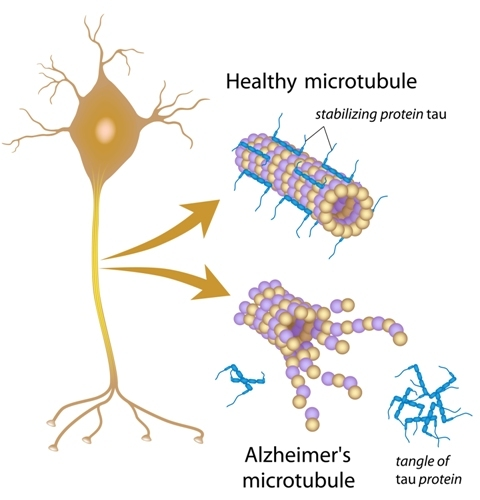
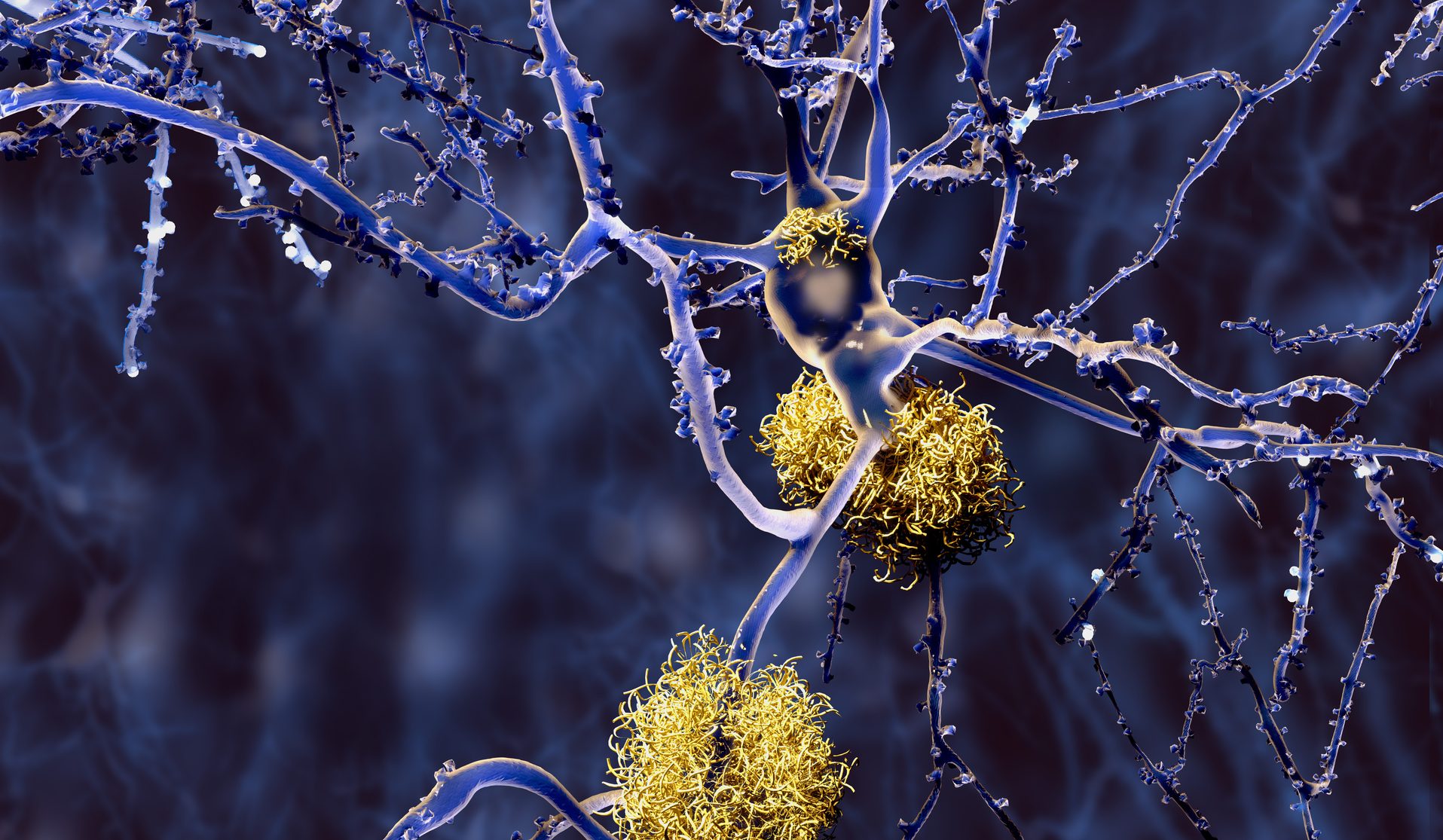
P-tau181 is a highly specific biomarker of AD and is a sub-type of misfolded tau protein. The occurrence of misfolded proteins can be triggered by genetics, environmental factors, or even head trauma to name a few. When a protein is misfolded it changes shape, leading to a functional change. Misfolded tau protein can also negatively change the shape of other correctly folded tau proteins, like a prion in Mad Cow disease, triggering neurofibrillary tangles (NFTs) to continue to aggregate and propagate down nerve networks interfering with neuronal functioning and causing cognitive decline in AD.
Blood levels of p-tau 181 can differentiate AD
from other neurodegenerative diseases, as well as predicting disease staging
and the rate of cognitive decline. Subjects with AD were compared with cognitively
unimpaired age-matched controls, patients with mild cognitive impairment (MCI),
those with frontotemporal dementia and other neurodegenerative disorders, as
well as healthy young adults. Established cerebrospinal fluid (CSF) and PET
biomarkers were collected to compare the capability of blood p-tau181 for identifying
AD.
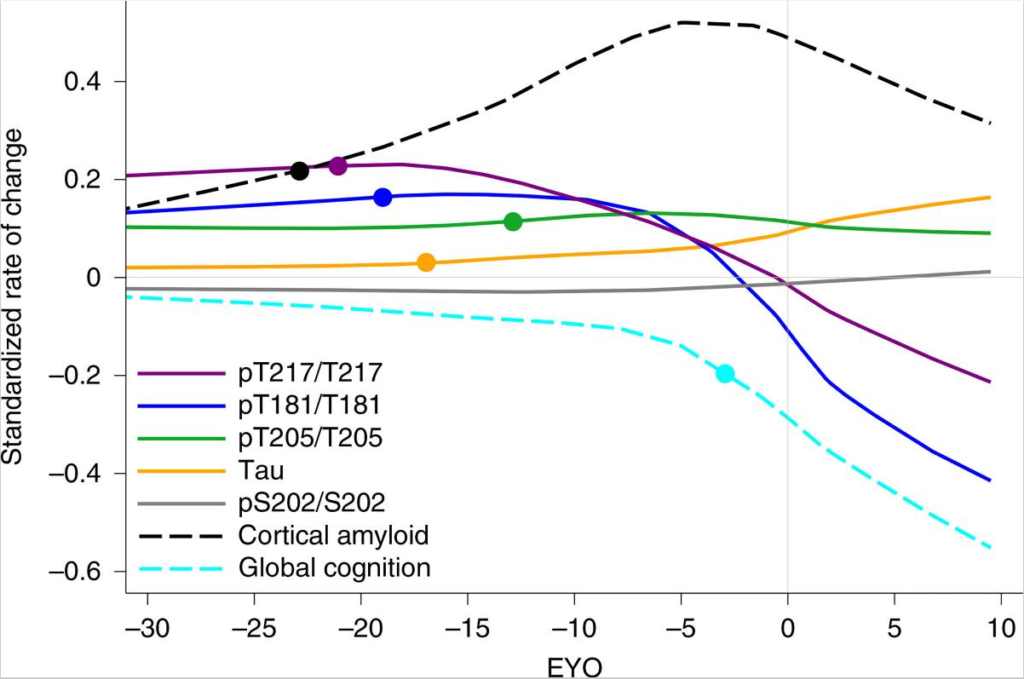
Concentrations of serum p-tau181 significantly increased with cognitive decline across groups. The lowest p-tau181 concentrations were found in healthy young adults and cognitively unimpaired older adults. The next highest levels were found in amyloid β-positive cognitively unimpaired older adults and those with MCI. The highest concentrations were found in amyloid β-positive AD patients. Serum p-tau181 was not only sensitive in correctly identifying AD stages but also specifically ruled out other causes of dementia.
A simple blood test would be invaluable for identifying and assessing AD in the community and clinical trials, especially since such p-tau181 concentrations correlate to AD risk. Here at the Center for Cognitive Health, we offer an AD prevention trial utilizing the p-tau 217 blood test, developed by Lilly, to assess the risk for developing AD in those with no memory problems–TRAILBLAZER-ALZ3 is using Donanemab (an antibody that targets amyloid-β), hopefully to prevent AD from developing. The days of needing a dose of radioactivity for an Amyloid PET scan or a spinal tap for CSF assessment may soon become obsolete. Hopefully, the results of this study will determine if treatment prevention (e.g., Donanemab) based on p-tau blood levels will be successful. If interested in knowing more about the study mentioned above, please visit our clinical trials page or give us a call at 503-207-2066 to find out more about disease modifying opportunities.