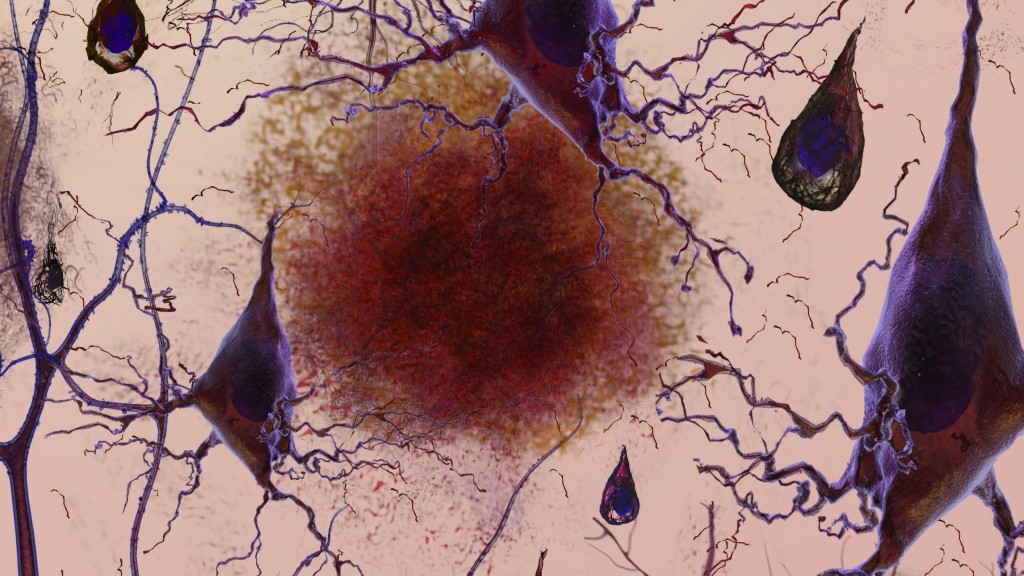
The amyloid hypothesis has inspired most of Alzheimer’s research for the last 20 years. It proposes that accumulation of a protein fragment known as beta-amyloid, and in particular the 42 amino acid subtype, is the underlying cause of the Alzheimer’s pathogenesis. It is a “sticky” compound that builds up within the brain 10-15 years before symptoms emerge. In theory, it disrupts synaptic communication, and ultimately ends in neuronal death by initiating neural fibrillary tangle (NFT) formation. The theory posits overproduction, poor disposal, and eventual accumulation of beta-amyloid is the primary cause of Alzheimer’s disease.
Beta-amyloid is only a small section of the larger amyloid precursor protein (APP). At present, scientists have not determined the normal function of APP. However, they know that when the protein is activated, it is divided into smaller pieces that are both inside or outside of cells. When pieces of beta-amyloid begin to accumulate, they form small clusters called oligomers. Multiple recent studies have implicated beta-amyloid oligomers as upstream drivers of Alzheimer’s disease. As these oligomers accumulate in the brain, they create chains of clusters known as fibrils, which then bond together to form beta-sheets. The characteristic amyloid plaques found in Alzheimer’s patients are composed of beta-sheets meshed together with other substances, such as tau protein making up NFTs. These plaques are believed to create intra-neuronal NFTs that cause synaptic injury, loss of neuronal function, and eventually cell death.
Genetic studies of familial Alzheimer’s patients identified over 200 mutations that lead to an early onset of the disease. Many of these mutations occur in the APP gene and cause an increase in the production of beta-amyloid. Overproduction causes an increase in misfolded beta-amyloid, resulting in the formation of amyloid oligomers and eventually plaques.
The APOE4 genotype has been identified as the greatest risk factor for developing Alzheimer’s disease. Those who carry the E4 allele of apolipoprotein E (APOE) have a much greater risk for developing early onset Alzheimer’s disease. The APOE4 genotype, which has been identified in approximately 65% of Alzheimer’s patients, is associated with reduced ability to clear amyloid from the brain and increased aggregation of beta-amyloid into oligomers.
Another argument for the amyloid hypothesis comes from examining the brains of individuals with Down syndrome. Adults with Down syndrome have three copies of the chromosome carrying the APP gene: chromosome 21. These individuals invariably develop beta-amyloid plaques identical to those seen in Alzheimer’s disease and have a high risk of developing early onset dementia. Thus, overproduction of beta-amyloid plaques in Down syndrome results from a comparable genetic pathology to Alzheimer’s patients with mutations of the APP gene.
Clinical trials of anti-amyloid agents that target beta-amyloid oligomers, such as aducanumab, donanemab, and lecanemab, have exhibited statistically significant clinical efficacy. Though as discussed in previous blog posts, the benefit received from these drugs is directly related to how early they’re given in the progression of the disease, generally before amyloid plaques have accumulated to the point of causing too much neural damage, which suggests that beta-amyloid accumulation initiates the cascade of Alzheimer’s pathogenesis.
The genetic and clinical trial data suggests beta-amyloid plays a key part in Alzheimer’s disease. The research sparked by the amyloid hypothesis remains relevant to this day and continues to inspire further clinical investigation. However, there is further emerging evidence that suggests it is far from the only factor influencing pathogenesis. Future studies targeting beta-amyloid and further examining APP mutations may one day lead to promising therapeutic interventions for Alzheimer’s disease, but alternative methods should also be pursued. Check in next week when we’ll be discussing these alternative methods.
Sources:
Annus, T., Wilson, L. R., Hong, Y. T., Acosta–Cabronero, J., Fryer, T. D., Cardenas–Blanco, A., Smith, R., Boros, I., Coles, J. P., Aigbirhio, F. I., Menon, D. K., Zaman, S. H., Nestor, P. J., & Holland, A. J. (2016). The pattern of amyloid accumulation in the brains of adults with Down syndrome. Alzheimer’s & Dementia, 12(5), 538–545. https://doi.org/10.1016/j.jalz.2015.07.490
Beta-amyloid and the Amyloid Hypothesis. https://www.alz.org/national/documents/topicsheet_betaamyloid.pdf
Tolar, M., Hey, J., Power, A., & Abushakra, S. (2021). Neurotoxic Soluble Amyloid Oligomers Drive Alzheimer’s Pathogenesis and Represent a Clinically Validated Target for Slowing Disease Progression. International Journal of Molecular Sciences, 22, 6355. https://doi.org/10.3390/ijms22126355