When our bodies are threatened by an invasive, harmful material – like a virus or a toxic chemical – our immune system responds quickly with inflammation. Neutrophils, macrophages, and lymphocytes are some of the most important cells involved in this process, allowing for regulation of the inflammatory response. This inflammatory response occurs in the brain, too, and is known as neuroinflammation. When neuroinflammation occurs, specialized cells in the brain known as astrocytes and microglia become activated, and inflammatory mediators are released.
When activated microglia release inflammatory mediators, the blood brain barrier is weakened, allowing immune cells to enter the central nervous system from the periphery of the body to assist with the inflammatory response. The balance of these mediators is crucial, as the immune system must respond to the bodily threat without causing enough inflammation to damage the brain. As such, these mediators – known as chemokines and cytokines – may be neuroprotective or neuroinflammatory. For example, a chemokine known as MCP-1 (monocyte chemoattractant protein-1) is responsible for regulating the migration of monocytes and T-cell lymphocytes toward the affected area, so it is neuroinflammatory. Conversely, activation of the GLP-1 (glucagon-like peptide 1) receptor shifts microglia from one subtype to another, inhibiting production of astrocytes and reducing inflammation.
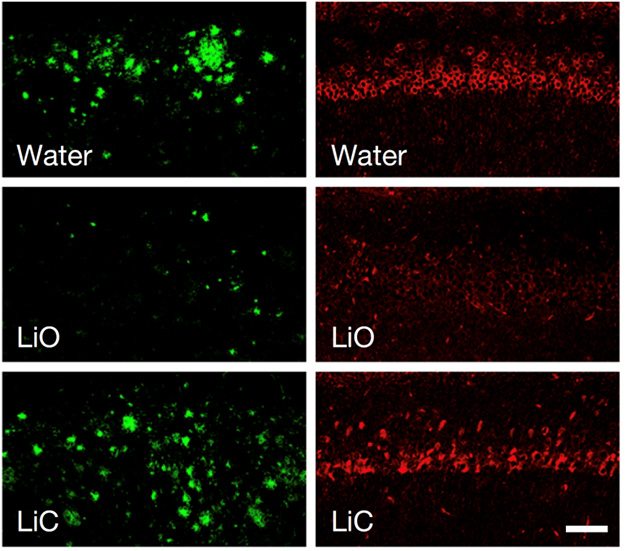
In Alzheimer’s disease, the accumulation of misfolded amyloid beta and tau proteins is associated with neuroinflammation. These misfolded proteins are perceived as harmful, thereby activating the inflammatory response. This creates a state of chronic inflammation, wherein the continuous inflammatory response begins to cause neuronal damage, contributing to the neurodegeneration associated with Alzheimer’s disease. For this reason, drugs that suppress the inflammatory response are being studied in Alzheimer’s disease.
A new study drug known as VHB9327 is being researched for its effects on TREM2, a receptor activated and upregulated in response to central nervous system injury. TREM2 allows microglia to transition to a specialized subtype known as disease-associated microglia. Disease-associated microglia emerge in response to neurodegenerative diseases, have a unique upregulation and downregulation of genes, and assist in the clearing of toxic aggregates of protein. Because TREM2 promotes disease-associated microglia, it is hypothesized that using VHB9327 to selectively activate TREM2 will skew microglia toward a neuroprotective, anti-inflammatory phenotype.
Preclinical studies in animal models have corroborated the above hypothesis, and VHB9327 is now being studied in clinical trials for Alzheimer’s disease. Key criteria for study eligibility includes a diagnosis of mild cognitive impairment or Alzheimer’s disease, a confirmation using cerebrospinal fluid or amyloid PET scan, and being between ages 50-85 at the start of the study. Exclusion criteria includes dementia for a reason other than AD, stroke in the last 12 months, use of anticoagulants, presence of cancer, or presence of any other neurological disease. The study will gather information about the effects of VHB937 on memory, cognition, daily activities, and brain imaging. Approximately 400 participants will receive either VHB9327 or placebo for 72 weeks. This is followed by an extension period where all participants will receive VHB9327. Publicly available information about the trial can be found here: https://clinicaltrials.gov/study/NCT07094516.