Microplastics and Nanoplastics (MNPs) are tiny pieces of plastic that are invisible to the naked eye, but have been found to exist in almost every environment imaginable. Microplastics are described as plastics measuring from 1 µm to 5 mm and Nanoplastics are described as any plastic piece smaller than 1 µm. These MNPs result from the breakdown of larger plastics. MNP levels in the brains of people with Alzheimer’s disease, vascular disease, and Parkinson’s disease appear to further increase inflammation, breakdown the blood brain barrier, and cause mitochondrial dysfunction leading to neuronal death. All MNP’s arrive in the brain from the gut-brain axis from the water and our foods.
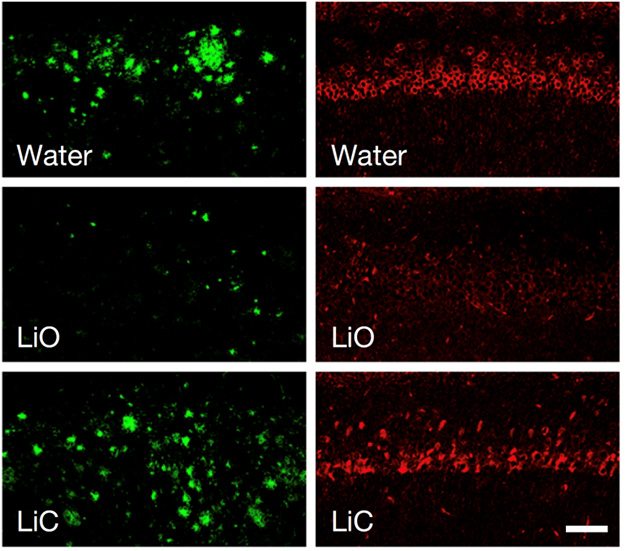
When measuring the levels of MNPs in human brains, livers, and kidneys, investigators unexpectedly found that levels of microplastics in the brain were much higher than in the liver or kidney. Also, the most common category of MNPs found in all three bodily regions was polyethylene. In the brain, nearly 75% of the MNPs that were found were polyethylene, which was a higher percentage than in the liver or kidney. The levels of microplastics in the frontal cortex of those individuals with diagnoses of dementia-causing diseases was found to be even greater. With certain individuals with dementia having 10 times more MNP accumulation than their cognitively normal counterparts. Why do MNPs concentrate more intensely in the brain?
Polyethylene is the most common plastic in the world. It is split into two major types: Low-Density Polyethylene and High-Density Polyethylene, which together, account for nearly all the single-use plastic in everyday life. If not properly handled and recycled, this plastic, along with other categories of plastic, are left to break down in oceans and landfills. Every piece of plastic that has ever been created still exists in our environment, in some form or another. When plastic is improperly disposed of, it is left to degrade into smaller pieces, eventually leading to MNPs. Although polyethylene is generally considered non-toxic, there is very little research into the effects of microplastics on our environment and in our bodies.
More research needs to be done to explore MNPs and to decipher the reason for the increase in MNP accumulation in the brain of people with dementia. One likely theory is that the increased MNP accumulation is the result of age-related increased impairment of normal brain mechanisms. Another, more worrying and less-likely theory, is that the increased MNP accumulation could be an early cause of decline in dementia-causing diseases. There is no current cause for alarm or panic, but it does pose several MNPs related questions. To minimize exposure to MNPs, limit the use of single-use plastics and properly recycle the plastics that require disposal.
Sources:
Dennis, J., Arulraj, D., & Mistri, T. K. (2025). Unseen toxins: Exploring the human health consequences of micro and nanoplastics. Toxicology reports, 14, 101955. https://doi.org/10.1016/j.toxrep.2025.101955
Gecegelen, E., Ucdal, M., & Dogu, B. B. (2025). A novel risk factor for dementia: chronic microplastic exposure. Frontiers in neurology, 16, 1581109. https://doi.org/10.3389/fneur.2025.1581109
Nihart, A.J., Garcia, M.A., El Hayek, E. et al. (2025). Bioaccumulation of microplastics in decedent human brains. Nat Med, 31, 1114–1119. https://doi.org/10.1038/s41591-024-03453-1
Valdivia, S., Riquelme, C., Carrasco, M. C., Weisser, P., Añazco, C., Alarcón, A., & Alarcón, S. (2025). Polyethylene Microplastics and Human Cells: A Critical Review. Toxics, 13(9), 756. https://doi.org/10.3390/toxics13090756