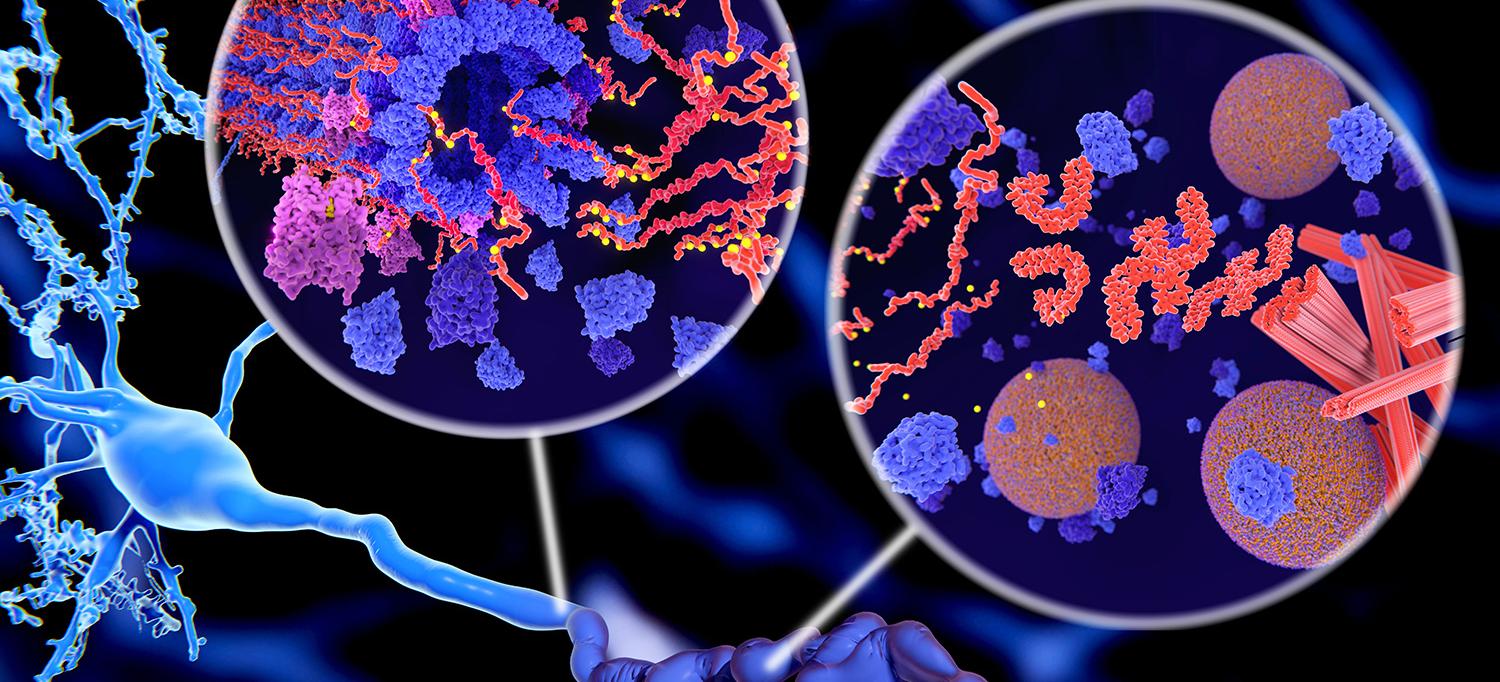
Memory impairment is often the hallmark symptom of Alzheimer’s disease (AD). However, research recently discovered a trend suggesting that decreased learning may preface memory loss in the preclinical phase of AD. Amyloid-positive patients in the preclinical stage of AD first experience a decline in learning ability while their memory is still comparable to amyloid-negative healthy controls. This discovery, much like the blood test we discussed a few blogs ago, may become a means of determining whether someone is going to develop AD well before memory impairment ensues.
During the preclinical stage of AD both amyloid-positive and amyloid-negative participants score equivalently on episodic memory tests. Over several years of repeat testing the amyloid-negative group improved their scores as a product of practice while amyloid-positive participants stagnated. This suggests that an impairment of learning precedes an impairment of episodic memory in the preclinical stage of AD.
Investigating further, scientists developed a test called the Online Repeated Cognitive Assessment (ORCA) where participants spend 30 minutes a day viewing Chinese characters paired with a correct or incorrect English word. After each session, patients reviewed their scores to see what pairings correctly matched, allowing them to learn from their previous mistakes. After several days participants began learning which Chinese characters represented the correct English words and which were incorrect. A previous study using this same paradigm found that amyloid-positive participants made more mistakes than the amyloid-negative group by the second session, and the gap continued to widen over the following days.
A more recent study tested 80 cognitively normal participants, 38 of whom were amyloid positive, on the ORCA along with other cognitive tests and neuroimaging. The amyloid-positive group showed learning deficits in the first session and the reduced learning compared to amyloid-negative individuals continued to grow over 6 days. After 6 days, the average ORCA scores of the two groups differed significantly, with amyloid-positive individuals showing reduced learning abilities compared to amyloid-negative patients. However, on episodic memory tests, both groups scored similarly, supporting the hypothesis that learning deficits may establish before memory impairment begins in amyloid-positive patients.
A well-designed learning test may be more useful for testing in the early stages of AD than the current standard measures of delayed recall. Generally, this type of “practice effect” learning, is avoided in clinical trials as it can confound memory test scores. Although, some experts who promote practice effect testing feel that decreased learning of new information echoes patient complaints of those that notice dysfunction but score normally on memory tests. This provides a means of testing the longitudinal progression of AD prior to the onset of memory impairment. Unfortunately, the test is not yet ready for use in clinical trials. Hopefully in the future this improved testing will translate to improved therapies for AD but only time will tell.