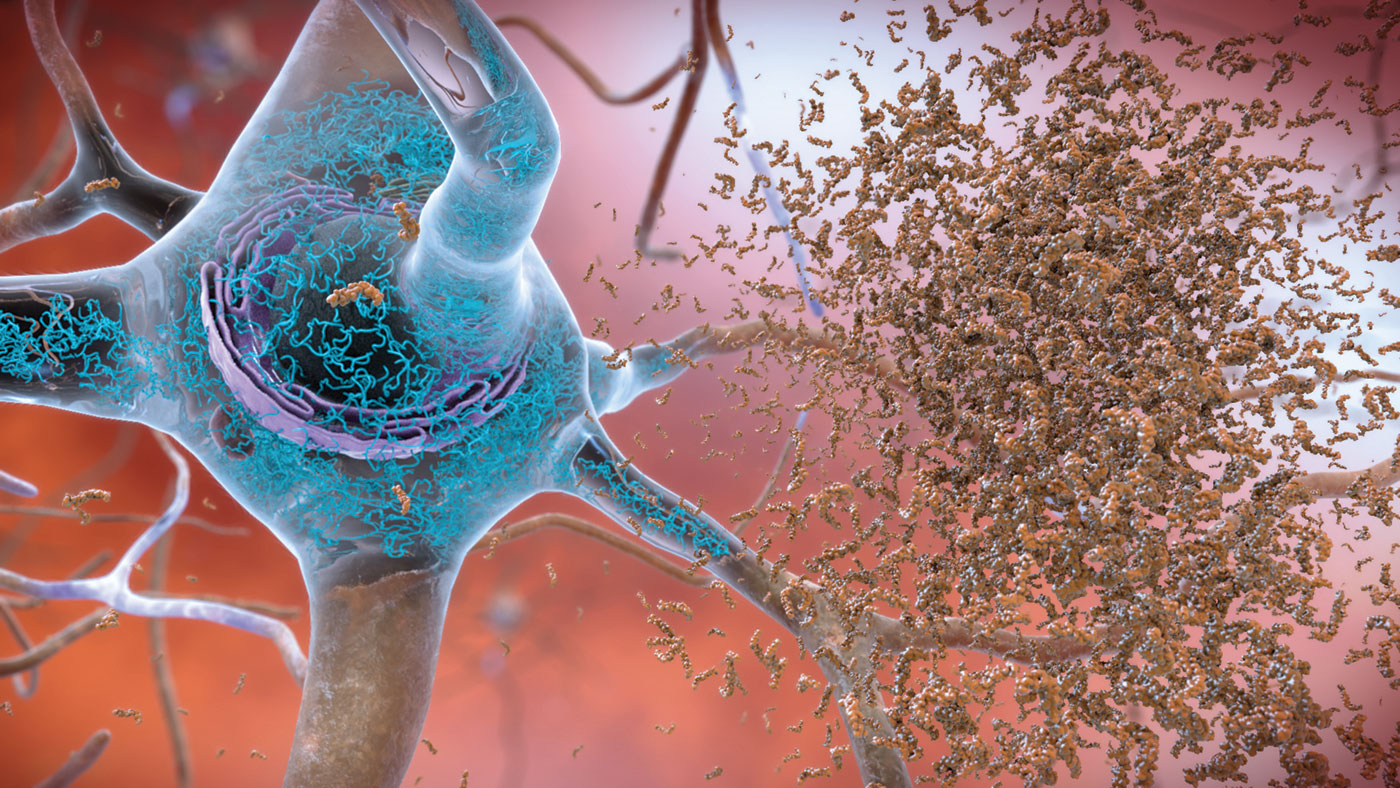
Alzheimer’s disease (AD) was identified in 1901 by Alois Alzheimer but despite being known for over a century, researchers are discovering new things about the disease today. For example, our regular readers should be familiar with the term “amyloid plaques” which are likely the most well researched biomarkers of AD. There are also sub-types of amyloid plaques and a group of pathologists recently observed what they believe to be a unique sub-type of plaque termed “coarse-grained plaques”. This week we will discuss these new plaques and their significance to AD research.
Coarse-grained plaques form a core of amyloid-ꞵ with 42 residues (Aꞵ42) surrounded by a shell of Aꞵ40, and are packed with a protein called norrin, a marker of blood-vessel damage. These plaques tend to localize near blood vessels but cannot cross the blood-brain barrier (BBB). 84% of the analyzed coarse-grained plaques had direct contact with vasculature, suggesting that they may induce cerebrovascular dysfunction rather than neurological dysfunction directly.
They are more common in people with two copies of the ApoE4 allele or those with early onset AD. In this study out of 28 non-ApoE4 carriers, 15 had sparse/frequent coarse-grained plaques, compared to 25 out of 33 for heterozygous ApoE4 carriers. All 11 homozygous carriers had moderate/frequent deposition of the coarse-grained plaques. This suggests interaction between the ApoE gene and coarse-grained plaques. In fact, there may even be a separate ApoE4-induced AD sub-type in homozygous carriers.
Researchers used laser scanning microscopy to visualize the plaques and saw that out of 74 brains, 38 had early onset AD, 21 had late onset, and 15 were never diagnosed with dementia though amyloid positive. This diverse grouping shows that, while coarse-grained plaques might induce early AD in many cases, it does not guarantee a specific pathological presentation. Staining techniques and complement testing were utilized to discover more about the plaques on a molecular level.
The stained coarse-grained plaques showed amyloid precursor protein (APP) and prion protein suggesting they may damage nearby neurons. The complement testing also revealed that these plaques have markers for extreme neuroinflammation and presence of astrocytes and microglia. In fact, microglia and astrocytes cover these plaques in a particular pattern which seems to further differentiate this type of plaque from those which were known previously, confirming the unique nature of these new plaques.
In summary, these specific plaques are only just beginning to be researched but already it seems there is a close relationship between the presence of coarse-grained plaques, the ApoE4 allele, cerebrovasculature, and AD pathology. With more studies and larger sample sizes, research of this topic may lead to innovation in the diagnosis and differing treatment options for subtypes of AD.